Bethe-Salpeter on top of quasiparticle energies: Difference between revisions
| Line 40: | Line 40: | ||
plot 'o-3D_QP_BSE.eps_q1_diago_bse' u 1:2 w l t 'Explicit QP', 'o-3D_BSE.eps_q1_diago_bse' u 1:2 w l t 'Scissor' | plot 'o-3D_QP_BSE.eps_q1_diago_bse' u 1:2 w l t 'Explicit QP', 'o-3D_BSE.eps_q1_diago_bse' u 1:2 w l t 'Scissor' | ||
[[File:03 bse diago qp.png|none|600px]] | [[File:03 bse diago qp.png|none|600px]] | ||
It is clear that this makes a difference in the peak distribution and intensity. Note that beside a simple shift you can renormalise as well the bandwidth of the valence and conduction bands in <code>KfnQP_E</code> (respectively the third and second value). You can try as an exercise to set up a new calculation using e.g. <code>1. | It is clear that this makes a difference in the peak distribution and intensity. Note that beside a simple shift you can renormalise as well the bandwidth of the valence and conduction bands in <code>KfnQP_E</code> (respectively the third and second value). You can try as an exercise to set up a new calculation using e.g. <code> 1.431042 | 1.200000 | 0.900000 |</code> for <code>KfnQP_E</code>. | ||
==Summary== | ==Summary== | ||
Revision as of 09:25, 25 March 2021
In this module you will learn how to include GW-corrected (QP) energies in the calculation of the absorption spectrum.
Prerequisites
Previous modules
- You must have completed the BSE kernel tutorial
- Optionally, also the absorption spectrum tutorial without QP corrections.
You will need:
- The
SAVEdatabases for 3D hBN - The
ndb.BS_Q1_CPU_0andndb.em1s*databases for 3D hBN - The
ndb.QPdatabase containing the GW-corrected band energies yamboexecutable
Reading the QP corrections from a previous GW calculation
In this previous calculation we have used a simple scissor operator to correct the Kohn-Sam DFT energies. In this part we see how we can instead take the corrections from a previous Yambo Gw calculation. We create and edit the input:
$yambo -F 03_3D_QP_BSE.in -y d -V qp -J 3D_BSE
We set all parameters as in the previous calculation, except for the part regarding the QP correction:
KfnQPdb= "E < 3D_QP_BSE/ndb.QP" # [EXTQP BSK BSS] Database KfnQP_N= 1 # [EXTQP BSK BSS] Interpolation neighbours % KfnQP_E 0.000000 | 1.000000 | 1.000000 | # [EXTQP BSK BSS] E parameters (c/v) eV|adim|adim %
Instead of setting the values for the scissor, we give the path to a database (3D_QP_BSE/ndb.QP) which contains the QP corrections. This has been created by running a GW calculation as in the GW tutorial. Run Yambo:
$ yambo -F 03_3D_QP_BSE.in -J "3D_QP_BSE,3D_BSE"
Note how we specified two directories, separated by a comma. This instructs yambo to look for databases which may be already present from previous steps of the calculations in all the listed folders. If you completed the previous section of the tutorial, then you should have the 3D_BSE<\code> folder with ndb.dipoles_*, ndb.em1s* and ndb.BS_CPU* databases, which will not be recalculated by yambo: the code will repeat only the diagonalization step, and print the resulting databases in the new folder 3D_QP_BSE. If you don't have the 3D_BSE folder, you have to repeat the entire BSE calculation, therefore you just need to give the option "-J 3D_QP_BSE".
This produces the following log in the standard output. Note Section 4 (regarding external QP corrections to the kernel):
<---> [04] External/Internal QP corrections
<---> E<3D_QP_BSE/ndb.QP[ PPA@E 27.21138 = XG 5 = Xb 1-40 = Scb 1-40]
<---> [dE_from_DB-Nearest K] Exact matches : 100.0000 [o/o]
This tells you that the file was found, read correctly and that the k points found in the file matched the ones you are using for the current calculation (otherwise interpolation would be needed).
It is crucial to check that the file has been read, since if not Yambo gives a warning but continues the calculation (with no QP corrections at all!).
As in the calculation with the scissor shift the final results are the files with the spectral functions. Let's compare the results for the optical absorption spectrum with those obtained previously with a simple scissor:
$ gnuplot
...
plot 'o-3D_QP_BSE.eps_q1_diago_bse' u 1:2 w l t 'Explicit QP', 'o-3D_BSE.eps_q1_diago_bse' u 1:2 w l t 'Scissor'
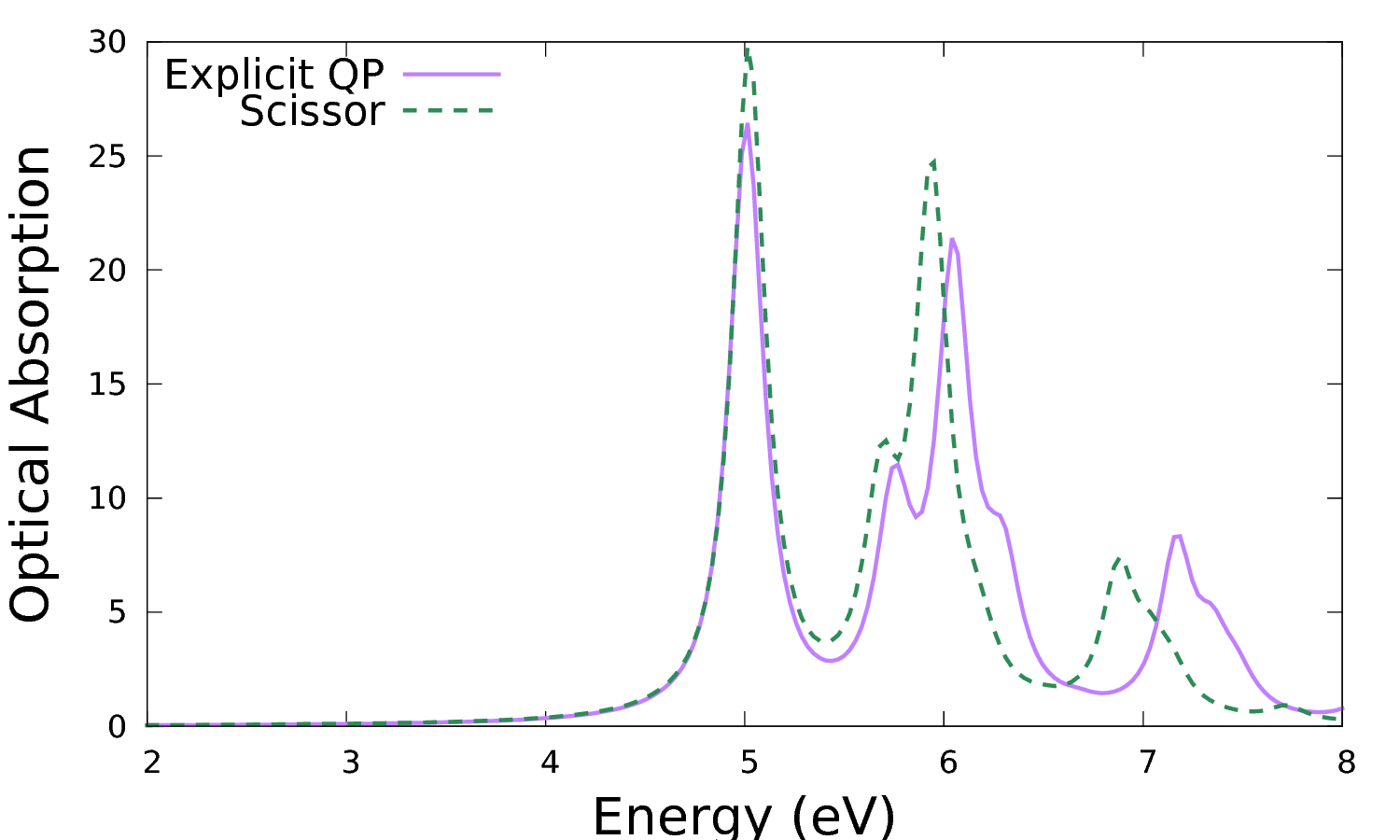

It is clear that this makes a difference in the peak distribution and intensity. Note that beside a simple shift you can renormalise as well the bandwidth of the valence and conduction bands in KfnQP_E (respectively the third and second value). You can try as an exercise to set up a new calculation using e.g. 1.431042 | 1.200000 | 0.900000 | for KfnQP_E.
Summary
From this tutorial you've learned:
- How to compute the optical spectrum including quasiparticle corrections (full GW+BSE calculation).
Links